

Huỳnh Lê Ngọc Long, Đinh Thuận Thiên, Học viên Cao học Lê Đào Hoàng Anh, PGS.TS. Trần Văn Hiếu
Nhóm nghiên cứu Y sinh học GMIF; PTN. Cảm biến sinh học, Khoa Sinh học-Công nghệ Sinh học, Trường Đại học Khoa học Tự nhiên, ĐHQG-HCM
---------
Hội chứng suy đường hô hấp cấp tính nghiêm trọng do virus SARS-CoV-2, có tên gọi chính thức là COVID-19, được ghi nhận xuất hiện lần đầu tiên ở Vũ Hán, Trung Quốc vào tháng 12/2019 và ca nhiễm đầu tiên được ghi nhận ở Việt Nam vào ngày 24/01/2020. Do sự lây lan nhanh chóng của virus trên toàn cầu, Tổ chức Y tế thế giới (World Health Organization-WHO) vào ngày 11/3/2020 đã tuyên bố COVID-19 là đại dịch toàn cầu [1]. Đến nay, số ca nhiễm COVID-19 lên tới hơn 200.000.000 và tử vong hơn 4.000.000 ca; trong đó, Việt Nam ghi nhận hơn 200.000 ca nhiễm và hơn 3.000 ca tử vong (được cập nhật vào ngày 10/8/2021) [2, 3]. Do hiện nay vẫn chưa có thuốc đặc trị COVID-19 nên vaccine vẫn là phương pháp chủng ngừa tiềm năng để khống chế đại dịch này. Các nghiên cứu về cơ chế xâm nhiễm của virus SARS-CoV-2 vào cơ thể vật chủ là nền tảng để nghiên cứu và phát triển vaccine phòng ngừa COVID-19.
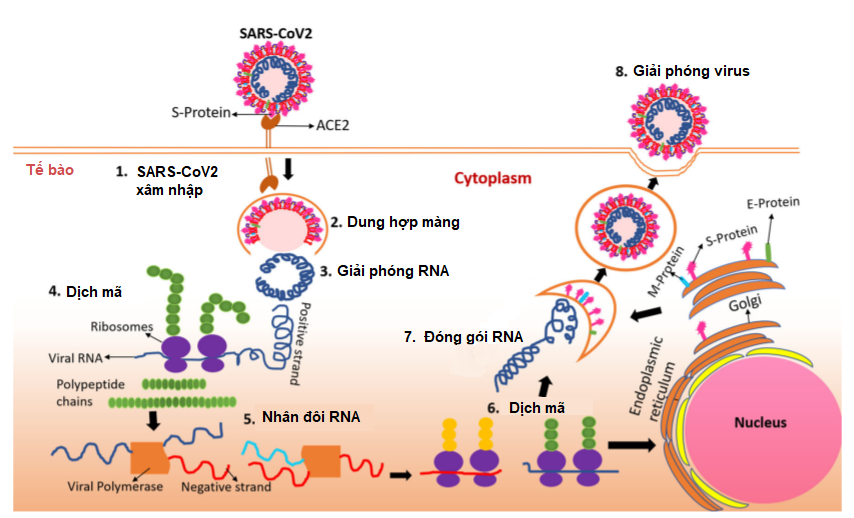
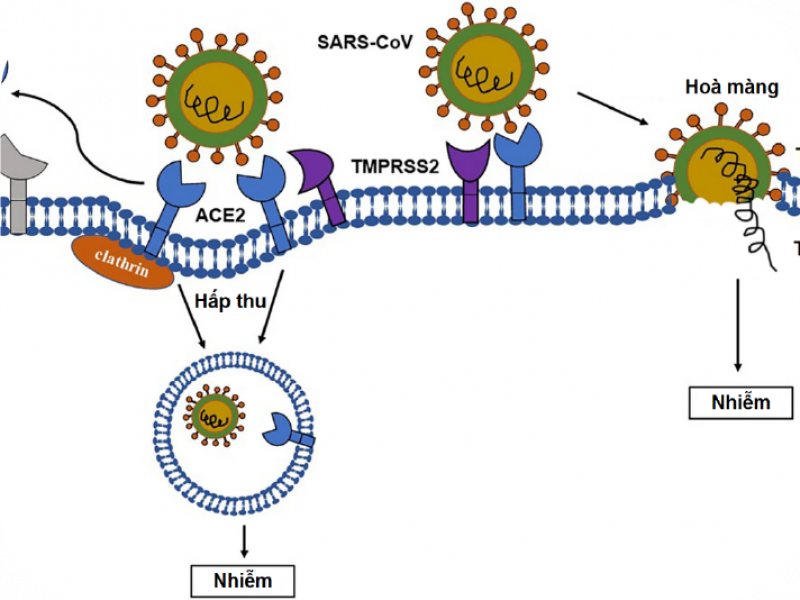
Vỏ virus SARS-CoV-2 có các protein S đính vỏ, bao gồm hai tiểu phần là S1 và S2; trong đó, tiểu phần S1 có vùng RBD (receptor binding domain - vùng gắn thụ thể) có chức năng tương tác với thụ thể ACE2 (angiotensin-converting enzyme 2 - ACE2) để xâm nhiễm vào cơ thể chủ; do vậy ACE2 ở người được coi là cánh cửa để virus xâm nhập vào tế bào (Hình 1) [4, 5]. Các nghiên cứu so sánh giữa virus SARS-CoV-2 và SARS-CoV (gây bệnh SARS) cho thấy sự liên kết ái lực cao hơn giữa virus SARS-CoV-2 và thụ thể ACE2 giúp virus SARS-CoV-2 dễ dàng nhập bào hơn so với SARS-CoV. Sau khi xâm nhiễm vào tế bào, virus sẽ sử dụng tế bào chủ để sản xuất các vật chất di truyền của nó; bên cạnh đó, chúng còn lẩn trốn hệ miễn dịch bằng cách bao phủ bề mặt protein S bằng các glycans có nguồn gốc từ tế bào chủ. Đồng thời, virus còn ngăn các tế bào bị xâm nhiễm truyền tín hiệu nhằm tạo thời gian để virus có thể nhân bản và lây nhiễm những tế bào lân cận. Các cơ chế này giúp virus SARS-CoV-2 vẫn có thể nhân bản và lây nhiễm trong cơ thể vật chủ chưa có biểu hiện bệnh. Ngoài ra, thụ thể ACE2 không chỉ có ở phổi mà còn ở các cơ quan khác như tim, thận, mạch máu, gan và hệ thần kinh trung ương; do đó, virus sau khi xâm nhiễm vào cơ thể thông qua đường hô hấp, chúng có thể tấn công vào bất kỳ cơ quan nào.
Các nhà khoa học hiện nay đang hướng đến việc thiết kế dạng vaccine chỉ là một phần cấu trúc của phân tử protein S chứa các epitope có thể kích hoạt được đáp ứng miễn dịch hiệu quả ở người. Tuy nhiên, điều đó không dễ dàng vì cấu trúc của protein S không chỉ toàn là amino acid mà chúng còn được đính thêm các nhóm đường (glycosylated) hoặc các nhóm acid béo (pamitoylated) [7]. Vì vậy, không phải bất cứ phần cấu trúc nào của protein S đều có thể được chọn để phát triển vaccine vì sự gắn của kháng thể có thể bị cản trở bởi các nhóm đường hoặc nhóm acid béo này. Để giải quyết bài toán này, Sikora và cộng sự (2021) đã sử dụng máy tính để mô phỏng chuyển động phân tử trên mô hình gồm bốn phân tử protein S có gắn các nhóm đường (glycosylated) được đính trên vỏ của virus trong môi trường nước nhằm tìm ra các vùng epitope tiềm năng cho việc thiết kế vaccine [2].
Các nhà khoa học sử dụng bốn tham số để đánh giá các vùng cấu trúc mà kháng thể có thể bám được bao gồm: khả năng tiếp cận của kháng thể (accessibility), sự bền vững của cấu trúc protein S (rigidity), sự bảo tồn của trình tự (sequence conservation) và tính sinh miễn dịch của trình tự amino acid (sequence-based immunogenicity). Mặc dù các dữ liệu cấu trúc của protein S hiện nay có rất nhiều trên các ngân hàng dữ liệu cấu trúc, nhưng một số phần cấu trúc của protein S (chẳng hạn như phần thân protein S và phần cấu trúc xuyên màng) có độ phân giải thấp do sự giới hạn của công nghệ giải cấu trúc hiện tại. Do đó, Sikora và các cộng sự đã dự đoán mô hình phân tử hoàn chỉnh protein S của SARS-CoV-2 dựa trên cơ sở dữ liệu cấu trúc sẵn có (Hình 2).
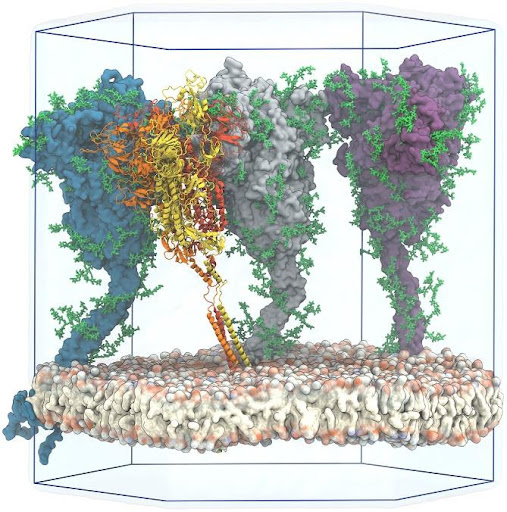
Sau khi đã có mô hình dự đoán hoàn chỉnh của protein S, các nhà khoa học sẽ kiểm tra sự bền vững cấu trúc (rigidity) của mô hình và phân tích khả năng tiếp cận không gian của kháng thể (accessibility) từ việc mô phỏng tương tác bám (docking) của protein S và vùng liên kết với kháng nguyên (Fab/antigen-binding fragment). Sau đó, mô phỏng chiếu chùm tia phân tán (diffuse ray) lên mô hình nhằm tìm ra các vị trí mà kháng thể có thể bám được. Cả hai mô hình có và không có nhóm đường đều được sử dụng trong quá trình mô phỏng này nhằm đánh giá ảnh hưởng của các nhóm đường đến khả năng bám của kháng thể.
Kết quả cho thấy rằng ở mô hình có nhóm đường có thể làm giảm khả năng tiếp cận của kháng thể đến từ 34% đến 80%. Tuy nhiên, các nhóm đường này không phải là nguyên nhân duy nhất làm giảm khả năng bám của kháng thể. Báo cáo thực nghiệm cho thấy rằng các protein S không chỉ phân bố rải rác trên vỏ virus mà chúng có thể kết nhóm và tương tác bám lẫn nhau [3]. Hiện tượng này có thể làm giảm khả năng bám của kháng thể đến khoảng từ 39% đến 86%.
Ngoài việc cho thấy ảnh hưởng của các nhóm đường lên khả năng bám của kháng thể, kết quả mô phỏng còn phát hiện ra các epitope sáng giá cho việc thiết kế vaccine. Tuy nhiên, một số epitope có thể chứa trình tự amino acid dễ đột biến hoặc không có khả năng sinh miễn dịch; nếu các vùng cấu trúc này được lựa chọn để thiết kế vaccine thì vaccine có nguy cơ sẽ không hiệu quả do virus có cơ hội đột biến ngay tại vùng cấu trúc tương ứng đó nhằm đào thoát miễn dịch. Vì vậy, Sikora và cộng sự kiểm tra sự bảo tồn của trình tự (sequence conservation) và tính sinh miễn dịch của trình tự amino acid (sequence-based immunogenicity) bằng các phần mềm ứng dụng tin sinh học. Sau khi tổng hợp các dữ liệu kết quả, nhóm nghiên cứu trên đã xác định được chín epitope nằm phần đỉnh của protein S. Đáng lưu ý, bốn trong số chín epitope được dự đoán bằng tin sinh học là epitope đã được biết từ các báo cáo thực nghiệm và lâm sàng. Điều này cho thấy phương pháp mô phỏng trên máy tính này có hiệu quả và đáng tin cậy trong việc dự đoán các epitope ứng cử viên cho thiết kế vaccine và phương pháp này có thể ứng dụng lên các cấu trúc protein S ở các virus gây bệnh khác.
Một nghiên cứu cho thấy rằng hầu hết các kháng thể đơn dòng (monoclonal antibodies - mAbs) tiềm năng có khả năng gắn tốt với vùng RBD trên protein S, qua đó ngăn cản sự gắn của protein S lên ACE2. Do đó, để tạo ra các kháng thể gắn RBD của protein S và trung hòa SARS-CoV-2, việc lập bản đồ các vị trí trên RBD tương tác với ACE2 là vô cùng quan trọng. Một nghiên cứu đã phân tích các vị trí gắn của kháng thể kháng SARS-CoV-2 bằng cách sử dụng các mô hình cấu trúc từ các cơ sở dữ liệu và 377 kháng thể đơn dòng thu được từ các bệnh nhân nhiễm bệnh. Kết quả cho thấy, 80 kháng thể được đánh giá là gắn được với vùng RBD và chỉ 19 trong số đó là có tiềm năng trung hòa virus SARS-CoV-2. Nếu mô tả cấu trúc vùng RBD như “phần thân người” thì các vị trí gắn thành có ở năm cụm: vai trái, cổ, vai phải đến sườn phải và một phần phân tán ở sườn trái (Hình 3). Khả năng trung hòa kháng thể từ epitope ở vùng cổ và vùng vai phải được đánh giá là cao nhất [9].
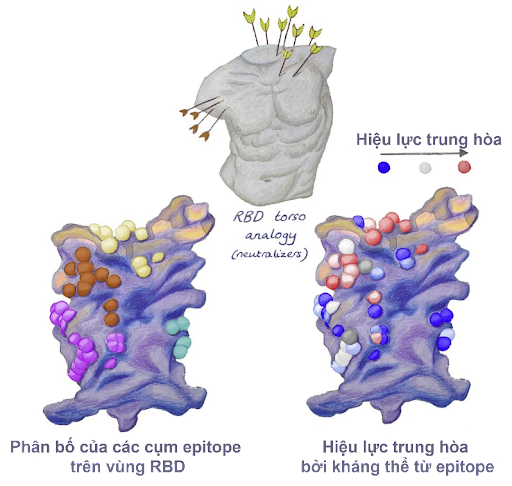
Nhờ có các nghiên cứu tin-sinh học trên máy tính đến thực nghiệm, chúng ta có thể dễ dàng thiết kế các kháng thể vừa có khả năng gắn vừa có khả năng trung hòa virus
SARS-CoV-2 để ngăn chặn xâm nhiễm. Ngoài ra, các kết quả còn gợi ý đến chiến lược phối hợp nhiều kháng thể trung hòa gắn ở nhiều cụm khác nhau để có thể bám các epitope trên protein S của vỏ virus ngăn cản sự tương tác với thụ thể ACE2. Đây có thể là liệu pháp miễn dịch với khả năng bảo vệ và chống lại tốt hơn với các biến thể của virus SARS-CoV-2 hiện nay. Từ đó cũng cho thấy sự hữu ích của phương pháp nghiên cứu trên máy tính (in silico) giúp phát triển vaccine chống lại đại dịch hiện nay và các bệnh khác trong tương lai.
Tài liệu tham khảo:
[1] W. H. Organization, "WHO Director-General's opening remarks at the media briefing on COVID-19 " 2020.
[2] W. H. Organization, "COVID-19 in Vietnam," 2021.
[3] "COVID-19 CORONAVIRUS PANDEMIC," 2021.
[4] C. O. Barnes et al., "Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies," Cell, vol. 182, no. 4, pp. 828-842.e16, 2020/08/20/ 2020.
[5] J. Shang et al., "Cell entry mechanisms of SARS-CoV-2," Proc Natl Acad Sci U S A, vol. 117, no. 21, pp. 11727-11734, May 26 2020.
[6] S. Boopathi, A. B. Poma, and P. Kolandaivel, "Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment," J Biomol Struct Dyn, vol. 39, no. 9, pp. 3409-3418, Jun 2021.
[7] Y. Watanabe, J. D. Allen, D. Wrapp, J. S. McLellan, and M. Crispin, "Site-specific glycan analysis of the SARS-CoV-2 spike," Science, vol. 369, no. 6501, pp. 330-333, Jul 17 2020.
[8] M. Sikora, S. von Bulow, F. E. C. Blanc, M. Gecht, R. Covino, and G. Hummer, "Computational epitope map of SARS-CoV-2 spike protein," PLoS Comput Biol, vol. 17, no. 4, p. e1008790, Apr 2021.
[9] W. Dejnirattisai et al., "The antigenic anatomy of SARS-CoV-2 receptor binding domain," Cell, vol. 184, no. 8, pp. 2183-2200.e22, 2021/04/15/ 2021.








Hãy là người bình luận đầu tiên