

Lê Quốc Tuấn, Trường Đại học Y Dược TP. HCM,
Nguyễn Thị Lệ, Trường Đại học Văn Lang,
Phùng Trung Hùng, Đại học Quốc gia TP. HCM
---------
Bệnh COVID-19 (Coronavirus disease 19) do virus SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) được ghi nhận lần đầu tiên tại Vũ Hán, Trung Quốc vào tháng 12 năm 2019, sau đó lây lan nhanh chóng thành đại dịch trên toàn thế giới [1]. Virus SARS-CoV-2 có vật chất di truyền là RNA sợi đơn và được phân loại vào nhóm beta coronavirus. Gai protein trên bề mặt virus (spike) bao gồm 2 tiểu đơn vị quan trọng, trong đó tiểu đơn vị S1 (receptor-binding subunit) chịu trách nhiệm gắn kết đặc hiệu lên men ACE2 của tế bào chủ (angiotensin-converting enzyme 2) và tiểu đơn vị S2 (membrane fusion subunit) chịu trách nhiệm hợp nhất màng virus vào màng tế bào chủ [2]. Sự xâm nhập của virus vào các phế bào làm kích hoạt các đại thực bào và tế bào đuôi gai, dẫn đến sự phóng thích của các hạt chứa yếu tố viêm (inflammasome) và kéo theo tình trạng độc tế bào tại chỗ [3]. Khi tình trạng độc tế bào gây ra bởi virus SARS-CoV-2 vẫn còn giới hạn trong lớp phế bào tại phổi thì bệnh COVID-19 vẫn chỉ biểu hiện dưới dạng viêm phổi với các mức độ từ nhẹ đến trung bình. Khi lớp nội mô mao mạch phổi xung quanh các phế nang bị tổn thương thì đáp ứng viêm toàn thân và tổn thương đa tạng mới xuất hiện như là dấu hiệu nặng của COVID-19 [4]. Như vậy, vấn đề điều chỉnh lại đáp ứng miễn dịch quá mức của cơ thể đối với SARS-CoV-2 chính là một ưu tiên hàng đầu trong các nghiên cứu về liệu pháp điều trị nhằm chống lại bệnh COVID-19 trong hoàn cảnh hiện nay [5]. Trong số các thuốc tác động vào hệ thống miễn dịch, corticosteroid là nhóm phổ biến nhất với nhiều kinh nghiệm trước đây trong điều trị các trường hợp viêm cấp tính và mạn tính, đồng thời hiệu quả và tác dụng phụ của corticosteroid cũng đã được giới y khoa biết đến rất rõ ràng và đầy đủ.
Biểu hiện lâm sàng của bệnh COVID-19
Đến nay, giới khoa học đã biết có 3 tác nhân thuộc gia đình coronavirus là SARS-CoV, SARS-CoV-2 và MERS-CoV có thể gây ra tử vong do viêm phổi trong một số ca nhiễm. Sau khi nhiễm SARS-CoV-2, các triệu chứng hô hấp sẽ xảy ra trong vòng 5-6 ngày sau khi tải lượng virus đạt đến đỉnh điểm và 97.5% bệnh nhân có triệu chứng tiếp tục diễn tiến thành bệnh COVID-19 trong vòng 2 tuần [5]. Tương tự như SARS-CoV và MERS-CoV, một nhóm nhỏ bệnh nhân nhiễm SARS-CoV-2 có biểu hiện khó thở và suy hô hấp nặng, thâm nhiễm phổi hai bên trên CT-Scan ngực, giảm lympho, đau cơ, đau khớp, đau đầu, chóng mặt, tiêu chảy, buồn nôn và ho ra máu [2-4]. Bệnh COVID-19 có thể thay đổi mức độ nặng từ viêm phổi đến phù phổi cấp và cả hội chứng ARDS (acute respiratory distress syndrome) với tình trạng đáp ứng viêm xảy ra mạnh mẽ [5].
Cách thức xâm nhập vào tế bào chủ của virus SARS-CoV-2
Về mặt di truyền (genomic), SARS-CoV-2 có sự tương đồng khoảng 80% so với SARS-CoV, khoảng 50% so với MERS-CoV, khoảng 90% so với cả bat-SL-CoVZC45 lẫn bat-SL-CoVZXC21. Đây là một bằng chứng gợi ý đến sự lây truyền virus từ dơi sang người lúc ban đầu [6]. Về mặt cấu trúc (proteomic), SARS-CoV-2 cũng có nhiều đặc điểm tương tự như SARS-CoV, trong đó cả hai loại virus này đều xâm nhập vào tế bào chủ bằng cách gắn các gai protein lên thụ thể là enzyme ACE2 (angiotensin-converting enzyme II) [7]. Ở người, quá trình xâm nhập của SARS-CoV-2 thông qua ACE2 phụ thuộc vào các enzyme TMPRSS2 (transmembrane serine protease 2) và CatB/L (endosomal cysteine proteases cathepsin B and L) vì đây là vị trí góp phần neo giữ các gai virus lại trên màng tế bào chủ [8]. Trong đó, enzyme TMPRSS2 được cho là có vai trò quan trọng hơn so với CatB/L. Vì vậy, phân tử ACE2 và TMPRSS2 có thể trở thành mục tiêu hàng đầu cho các liệu pháp điều trị COVID-19 trong tương lai.
Mặc dù SARS-CoV và SARS-CoV-2 có sự tương đồng về mặt di truyền và cấu trúc của vùng gắn kết thụ thể xâm nhập RBD (receptor-binding domain), tuy nhiên các kháng thể đơn dòng đối kháng với RBD không thể vô hiệu hóa được SARS-CoV-2 [9]. Điều này gợi ý rằng khả năng bảo vệ chéo cho hai loại virus này bị hạn chế. Mặt khác, ngoài con đường ACE2 thì SARS-CoV-2 còn có thể lây nhiễm vào các tế bào có biểu hiện thụ thể FcR (Fc receptors). Do đó, sau khi qua được phổi, sự xâm nhập virus vào tế bào chủ có thể xảy ra qua trung gian kháng thể tại đại thực bào, bạch cầu đơn nhân hoặc lympho B, ngay cả khi không có sự biểu hiện của ACE2 và TMPRSS2 trên các tế bào này [10].
Bằng chứng lâm sàng về đáp ứng viêm hệ thống quá mức trong COVID-19
Trên những bệnh nhân COVID-19 phụ thuộc oxy, nồng độ huyết tương của các protein phản ứng cấp tính, CRP và amyloid A được ghi nhận tăng lên, trong khi prealbumin có xu hướng giảm [11]. Tương tự, nồng độ của các cytokine viêm như IL-6, TNF-α, IL-1β, IL-2, IL-7 và IL-17 cũng gia tăng [12]. Bên cạnh đó, đáp ứng tiết IFN-I không đầy đủ cũng có thể góp phần vào các tổn thương phổi và đa cơ quan trong bệnh COVID-19 nặng [13].
Sự gia tăng các cytokine viêm đi đôi với mức độ nghiêm trọng của COVID-19 trên lâm sàng, nồng độ huyết tương của IL-6 và TNF-α càng cao thì nguy cơ tử vong càng lớn [14]. Đáp ứng miễn dịch của cơ thể đối với SARS-CoV-2 có vẻ phức tạp và không đồng nhất, một số bệnh nhân có sự kích hoạt và tăng sinh mạnh mẽ cả lympho T lẫn lympho B, trong khi khoảng 1/5 số bệnh nhân lại chỉ có đáp ứng miễn dịch ở mức thấp [15]. Có 3 mức độ đáp ứng miễn dịch đã được ghi nhận trên bệnh nhân COVID-19: (1) hoạt hóa mạnh mẽ lympho TCD4, (2) hoạt hóa nhẹ lympho TCD4 và (3) không phát hiện đáp ứng lympho bào [15]. Trong đó, mức độ đáp ứng miễn dịch kiểu 1 có liên quan đến các ca COVID-19 nặng hơn.
Cơ chế tăng đáp viêm quá mức và bão cytokine trong COVID-19
Khi nhiễm virus, các phế bào, đại thực bào và bạch cầu đơn nhân được kích hoạt tạo ra một lượng lớn các cytokine và chemokine, dẫn đến hóa ứng động thêm nhiều bạch cầu đơn nhân và lympho T. Bệnh nhân COVID-19 nặng có nồng độ cao của nhiều cytokine như IL-6, IL-2, IL-7, IL-10, G-CSF, TNF, CXCL10, MCP1 và MIP1α trong huyết thanh [4], có thể được gọi là hội chứng giải phóng cytokine CRS (cytokine release syndrome) hay bão cytokine [16]. Trong đó, nồng độ IL-6 trong máu cao hơn đáng kể ở những bệnh nhân tử vong [1, 13] và có thể dùng làm chỉ số dự đoán nhu cầu cần thở máy [17].
Như vậy, các giả thuyết đều cho rằng nguyên nhân chính gây tử vong ở COVID-19 là hội chứng ARDS với cơn bão cytokine đặc trưng. Các tổn thương đa cơ quan do đông máu nội mạch cũng gây ra bởi các cytokine nhất là IL-6 [18], được ghi nhận trên lâm sàng thông qua tình trạng giảm tiểu cầu và tăng D-dimer. Các vi huyết khối tại các tạng có liên quan chặt chẽ đến tiên lượng xấu và thường gặp tại phổi, chi dưới, tay, não, tim, gan và thận [19]. Một lý giải khác cho tổn thương đa cơ quan là hiện tượng nhiễm SARS-CoV-2 vào tế bào nội mô mạch máu, dẫn đến tăng tính thấm thành mạch và gây ra hiệu ứng ngộ độc tế bào [20].
Tín hiệu IL-6-STAT3 là nguyên nhân tiềm năng gây ra ARDS và tổn thương đa tạng
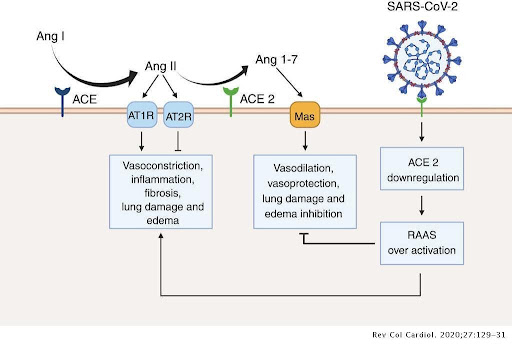
Nhiễm SARS-CoV-2 dẫn đến sự nhập bào của phức hợp men ACE2 và virus vào trong các phế bào và tế bào nội mô mao mạch phổi. Tình trạng này làm gia tăng nồng độ angiotensin II trong huyết tương do sự giảm biểu hiện của men ACE2 trên bề mặt tế bào [21]. Angiotensin II không chỉ hoạt động như một chất co mạch mà còn đóng vai trò là một cytokine viêm thông qua thụ thể AT1R (Angiotensin II type 1 receptor) [22]. Chính vì vậy, một giả thuyết mới được đưa ra rằng hệ thống RAS (renin-angiotensin system) có thể tham gia vào quá trình tiến triển thành ARDS sau khi bệnh nhân nhiễm SARS-CoV-2 [16]. Phức hợp angiotensin II - thụ thể AT1R kích hoạt lên enzyme ADAM17 (metallopeptidase domain 17), sau đó enzyme này sẽ chuyển đổi các yếu tố tăng trưởng biểu mô (EGF), TNF-α và IL-6Rα từ dạng kết dính trên màng thành dạng hoà tan, đưa đến hoạt hóa con đường NF-κB [16]. Do đó, angiotensin II và IL-6Rα hòa tan (sIL-6Rα) trong huyết tương có tiềm năng trở thành chất chỉ báo mức độ nặng của COVID-19.
Sau khi được tạo ra, phức hợp sIL-6R và IL-6 sẽ truyền tín hiệu vào nội bào bằng cách gắn lên phân tử gp130 nằm trên các tế bào không thuộc hệ miễn dịch như nội mô, biểu mô và nguyên bào sợi. Điều này đưa đến sự hoạt hóa hệ thống Janus kinase (JAK) / STAT3 [23]. Như vậy, thông qua trung gian IL-6, tín hiệu từ angiotensin II và AT1R có thể tạo ra một phản hồi ngược dương tính lên NF-κB và làm khuếch đại IL-6 (IL-6 amplifier), góp phần gây nên viêm phổi, ARDS, đông máu và suy đa cơ quan.
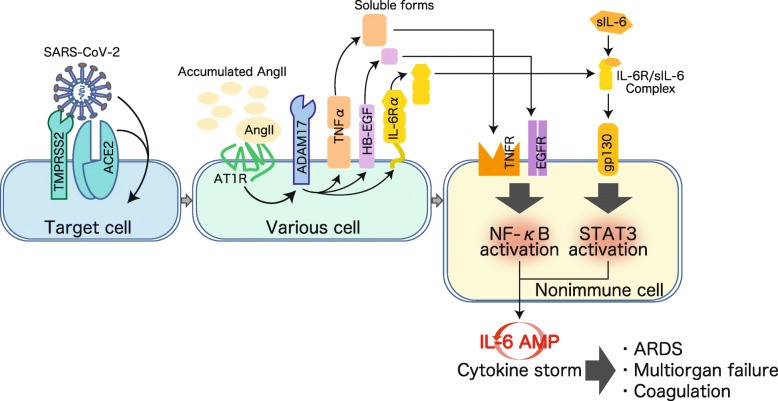
Hiện tượng khuếch đại IL-6 tại các tế bào không thuộc hệ miễn dịch được thực hiện bởi sự kích hoạt đồng thời hai yếu tố phiên mã NF-κB và STAT3. Các gen mục tiêu của NF-κB đã được ghi nhận bao gồm IL-6, chemokine và các yếu tố tăng trưởng [24]. Tuy nhiên, mức độ hoạt hóa của hệ thống IL-6 và NF-κB có sự khác biệt giữa các loại tế bào đặc hiệu, vì vậy mức độ của đáp ứng viêm tại các mô cũng có xu hướng khác nhau [24]. Sự hoạt hóa có xu hướng dễ xảy ra tại các tế bào đáy của khí quản, nguyên bào sợi, tế bào sừng, biểu mô ống thận và tế bào sụn. Ngoài ra, sự khuếch đại IL-6 còn phụ thuộc vào sự tương tác giữa các yếu tố môi trường và di truyền. Tình trạng stress, cảm giác đau và một số SNP được ghi nhận có thể ảnh hưởng đến sự khuếch đại IL-6 thông qua con đường NF-κB [25]. Tất cả những điều trên góp phần giải thích cho sự thay đổi biểu hiện của bệnh COVID-19 từ nhẹ đến nặng.
Corticosteroid ức chế đáp ứng viêm và cơn bão cytokine
Corticosteroid là một hoạt chất có hiệu quả kháng viêm đa dạng với các cơ chế tác động phức tạp cả trên gen (genomic) và ngoài gen (non-genomic), mang lại nhiều lợi ích trong quản lý các trường hợp nhiễm trùng nặng [26]. Trong cơ thể, lớp bó của tuyến vỏ thượng thận là nơi tiết ra hormon corticosteroid ở dạng glucocorticoid. Hoạt động kháng viêm của glucocorticoid thể hiện qua 2 cơ chế cơ bản: (1) kích thích tổng hợp và giải phóng các protein kháng viêm, và (2) ức chế các protein gây viêm. Glucocorticoid tan trong lipid và có thụ thể là phân tử GR (glucocorticoid receptor) nằm bên trong khu vực nội bào tại hầu hết các tế bào trong cơ thể. Khi gắn với glucocorticoid, thụ thể GR sẽ tách khỏi các protein Hsp70 (heat-shock proteins 70), Hsp90 và immunophilin [27]. Sau đó, phức hợp hormon và thụ thể GR này sẽ đi vào nhân tế bào và tương tác với các trình tự DNA đặc hiệu GRE (glucocorticoid responsive elements) trên vùng điều hòa của các gen đích [28]. Cuối cùng, glucocorticoid ngăn chặn sự biểu hiện của các gen gây viêm bằng cách ức chế enzyme histone acetyltransferase, đồng thời kích hoạt enzyme histone deacetylase.
Glucocorticoid có thể tác động lên nhiều nhóm tế bào không thuộc hệ miễn dịch đặc hiệu của cơ thể. Ví dụ như glucocorticoid sẽ ức chế lên sự biểu hiện của yếu tố phiên mã IRF3 (interferon regulatory factor 3), làm giảm sản xuất interferon [29]. Phức hợp GR-glucocorticoid cũng ức chế quá trình tạo ra các protein viêm bằng cách khóa yếu tố NF-κB thông qua việc làm tăng biểu hiện của protein ức chế IκBα [30]. Hoặc glucocorticoid kích thích sản xuất annexin 1, thông qua đó ức chế sự biểu hiện của enzyme phospholipase A2 [31]. Đây là những bằng chứng về cơ chế của các lộ trình tín hiệu nội bào liên quan đến vấn đề ức chế đáp ứng viêm bởi corticosteroid.
Glucocorticoid đóng vai trò quan trọng trong việc ức chế hoạt động của các bạch cầu. Glucocorticoid ngăn chặn sự sản xuất các chất đáp ứng viêm cấp tính và các chemokine [31], do đó làm giảm bớt phản ứng huy động bạch cầu. Glucocorticoid cũng làm giảm sự biểu hiện của các phân tử kết dính bạch cầu trên các tế bào chủ như ELAM-1 (endothelial-leukocyte adhesion molecule 1), ICAM-1 (intracellular adhesion molecule) và VCAM-1 (vascular adhesion molecule 1) [32]. Glucocorticoid có khả năng ức chế sự trưởng thành, biệt hóa và tăng sinh của tất cả các loại bạch cầu bao gồm 2 nhóm chính: (1) tế bào dòng tủy (như đại thực bào, bạch cầu đơn nhân, thực bào tại mô, tế bào đuôi gai, bạch cầu hạt) và (2) tế bào dòng lympho (như CD8, Th1, Th2 và B) [33]. Glucocorticoid cũng làm giảm sự biểu hiện của các phân tử MHC (major-histocompatibility-complex) và các thụ thể Fc [34] trên bề mặt màng tế bào chủ, do đó ngăn chặn sự trình diện kháng nguyên đối với các lympho T [35]. Bên cạnh đó, glucocorticoid còn ức chế sự hoạt hóa, tăng sinh và giải phóng các globulin miễn dịch từ lympho B [36], cũng như kích hoạt quá trình chết theo chương trình của lympho T và các tế bào đệm trong tuyến ức [37]. Các cơ chế này đều đưa đến kết quả làm giảm sự xuất hiện của cơn bão cytokine tại các mô trên lâm sàng.
Khuyến nghị corticosteroid trong thực hành điều trị COVID-19
Tương tự như nhiễm trùng huyết, ở các bệnh nhân COVID-19, hiện tượng mất điều hòa phản miễn dịch hệ thống của vật chủ là lý do dẫn đến bệnh cảnh tổn thương đa cơ quan nghiêm trọng. Corticosteroid là nhóm thuốc điều hòa miễn dịch mạnh và đa cơ chế, có thể giúp ngăn ngừa hoặc làm giảm đáp ứng viêm toàn thân quá mức trong các ca COVID-19 nặng. Dữ liệu từ các bằng chứng khoa học y sinh, các thử nghiệm ngẫu nhiên có đối chứng và các phân tích tổng hợp đều ủng hộ mạnh mẽ việc sử dụng corticosteroid cho các bệnh nhân COVID-19 cần hỗ trợ oxy. Theo khuyến cáo từ WHO 2020, những bệnh nhân không triệu chứng hoặc triệu chứng nhẹ không cần bổ sung oxy thì không nên sử dụng corticosteroid [38]. Các loại corticosteroid (như dexamethasone, hydrocortisone hoặc methylprednisolone) có thể được sử dụng qua tiêm tĩnh mạch hoặc đường uống, với liều tương đương 6mg dexamethasone trong thời gian từ 5 đến 10 ngày, tùy theo sự đánh giá mức độ tiến triển nặng của bệnh nhân COVID-19.
Tài liệu tham khảo
- Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062.
- Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620–2629.
- Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–513.
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506.
- Kim JY, Ko JH, Kim Y, Kim YJ, Kim JM, Chung YS, et al. Viral load kinetics of SARS-CoV-2 infection in first two patients in Korea. J Korean Med Sci. 2020;35(7):e86.
- Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574.
- Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–879.
- Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.
- Iwasaki A, Yang Y. The potential danger of suboptimal antibody responses in COVID-19. Nat Rev Immunol. 2020;20(6):339–341.
- Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63(3):364–374.
- Pan Y, Zhang D, Yang P, Poon LLM, Wang Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect Dis. 2020;20(4):411–412.
- Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun. 2020;111:102452.
- Mahmudpour M, Roozbeh J, Keshavarz M, Farrokhi S, Nabipour I. COVID-19 cytokine storm: the anger of inflammation. Cytokine. 2020;133:155151.
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454.
- Hirano T, Murakami M. COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity. 2020;52(5):731–733.
- Herold T, Jurinovic V, Arnreich C, Lipworth BJ, Hellmuth JC, von Bergwelt-Baildon M, et al. Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19. J Allergy Clin Immunol. 2020;146(1):128–136.
- Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355–362.
- Liu PP, Blet A, Smyth D, Li H. The science underlying COVID-19: implications for the cardiovascular system. Circulation. 2020;142(1):68–78.
- Park WB, Kwon NJ, Choi SJ, Kang CK, Choe PG, Kim JY, et al. Virus isolation from the first patient with SARS-CoV-2 in Korea. J Korean Med Sci. 2020;35(7):e84.
- Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–879.
- Eguchi S, Kawai T, Scalia R, Rizzo V. Understanding angiotensin II type 1 receptor signaling in vascular pathophysiology. Hypertension. 2018;71(5):804–810.
- Murakami M, Kamimura D, Hirano T. Pleiotropy and specificity: insights from the interleukin 6 family of cytokines. Immunity. 2019;50(4):812–831.
- Fujita M, Yamamoto Y, Jiang JJ, Atsumi T, Tanaka Y, Ohki T, et al. NEDD4 is involved in inflammation development during keloid formation. J Invest Dermatol. 2019;139(2):333–341.
- Murakami M, Harada M, Kamimura D, Ogura H, Okuyama Y, Kumai N, et al. Disease-association analysis of an inflammation-related feedback loop. Cell Rep. 2013;3(3):946–959.
- N Heming, S Sivanandamoorthy, P Meng, R Bounab, D. Annane. Immune effects of corticosteroids in sepsis. Front Immunol, 9 (2018), p. 1736.
- D Picard, B Khursheed, MJ Garabedian, MG Fortin, S Lindquist, KR. Yamamoto. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature, 348 (6297) (1990), pp. 166-168.
- RM Nissen, KR. Yamamoto. The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev, 14 (18) (2000), pp. 2314-2329.
- MM Reily, C Pantoja, X Hu, Y Chinenov, I. Rogatsky. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J, 25 (1) (2006), pp. 108-117.
- JP Vago, CR Nogueira, LP Tavares, FM Soriani, F Lopes, RC Russo, et al. Annexin a1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol, 92 (2) (2012), pp. 249-258.
- M Miyamasu, Y Misaki, S Izumi, T Takaishi, Y Morita, H Nakamura, et al. Glucocorticoids inhibit chemokine generation by human eosinophils. J Allergy Clin Immunol, 101 (1) (1998), pp. 75-83.
- BN Cronstein, SC Kimmel, RI Levin, F Martiniuk, G. Weissmann. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci U S A., 89 (21) (1992), pp. 9991-9995.
- LI McKay, JA. Cidlowski. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev, 20 (4) (1999), pp. 435-459.
- M Bianchi, C Meng, LB. Ivashkiv. Inhibition of IL-2-induced JAK-STAT signaling by glucocorticoids. Proc Natl Acad Sci U S A, 97 (17) (2000), pp. 9573-9578.
- RH DeKruyff, Y Fang, DT. Umetsu. Corticosteroids enhance the capacity of macrophages to induce Th2 cytokine synthesis in CD4+ lymphocytes by inhibiting IL-12 production. J Immunol, 160 (5) (1998), pp. 2231-2237.
- TR Cupps, TL Gerrard, RJ Falkoff, G Whalen, AS. Fauci. Effects of in vitro corticosteroids on B cell activation, proliferation, and differentiation. J Clin Invest, 75 (2) (1985), pp. 754-761.
- E van Vliet, M Melis, W. van Ewijk. The influence of dexamethasone treatment on the lymphoid and stromal composition of the mouse thymus: a flowcytometric and immunohistological analysis. Cell Immunol, 103 (2) (1986), pp. 229-240.
- World Health Organization 2020. Corticosteroids for COVID-19, Living Guidance, COVID-19: Clinical care







Hãy là người bình luận đầu tiên